Les médicaments biosimilaires sont issus d’une source biologique et sont développés par génie génétique en comparaison à un médicament biologique de référence déjà commercialisé et dont le brevet est tombé dans le domaine public. Un médicament biosimilaire est avant tout un médicament biologique1, ce n’est donc pas comme un médicament générique produit par synthèse chimique.
L’AMM d’un médicament biosimilaire est délivrée sur la base d’une équivalence des résultats thérapeutiques sur le plan chimique, clinique et biologique, et pas uniquement sur le plan chimique et sur la base de la bioéquivalence comme pour les médicaments génériques.
La démonstration de la similarité d’un médicament biosimilaire par raport à un médicament de référence nécessite des essais précliniques et cliniques, notamment en comparant la pharmacocinétique et en évaluant le rapport efficacité-tolérance avec le médicament biologique de référence.
Un médicament générique est défini d’après l’article L5121-1 du code de la santé publique comme étant un médicament ayant la même composition qualitative et quantitative en principe actif, la même forme pharmaceutique qu’une spécialité de référence, et dont la bioéquivalence avec la spécialité de référence est démontrée par des études de biodisponibilité appropriées.
Les médicaments génériques sont soumis aux mêmes exigences que le “médicament” princeps en termes de sécurité et d’efficacité (article L5121-1 du Code de la Santé Publique). L’obtention de leur AMM repose notamment sur la démonstration de la bioéquivalence avec le médicament princeps.
À la différence d’un médicament biosimilaire, un médicament générique ne nécessite pas de développement en recherche fondamentale (pas d’analyse physicochimique de biosimilarité ni d’études cliniques d’efficacité et de sécurité).

Les étapes de recherche et de fabrication durent près de 10 ans. Des études de comparaison par rapport au biomédicament de référence doivent être effectuées par les laboratoires pharmaceutiques. Pour être commercialisé, le biosimilaire est évalué de manière très stricte par l’Agence Européenne du Médicament (EMA). Leur efficacité et leur sécurité sont donc démontrées et sont similaires à leur médicament de référence. Vous serez donc traité de la même manière qu’avec le médicament de référence et avec le même résultat.
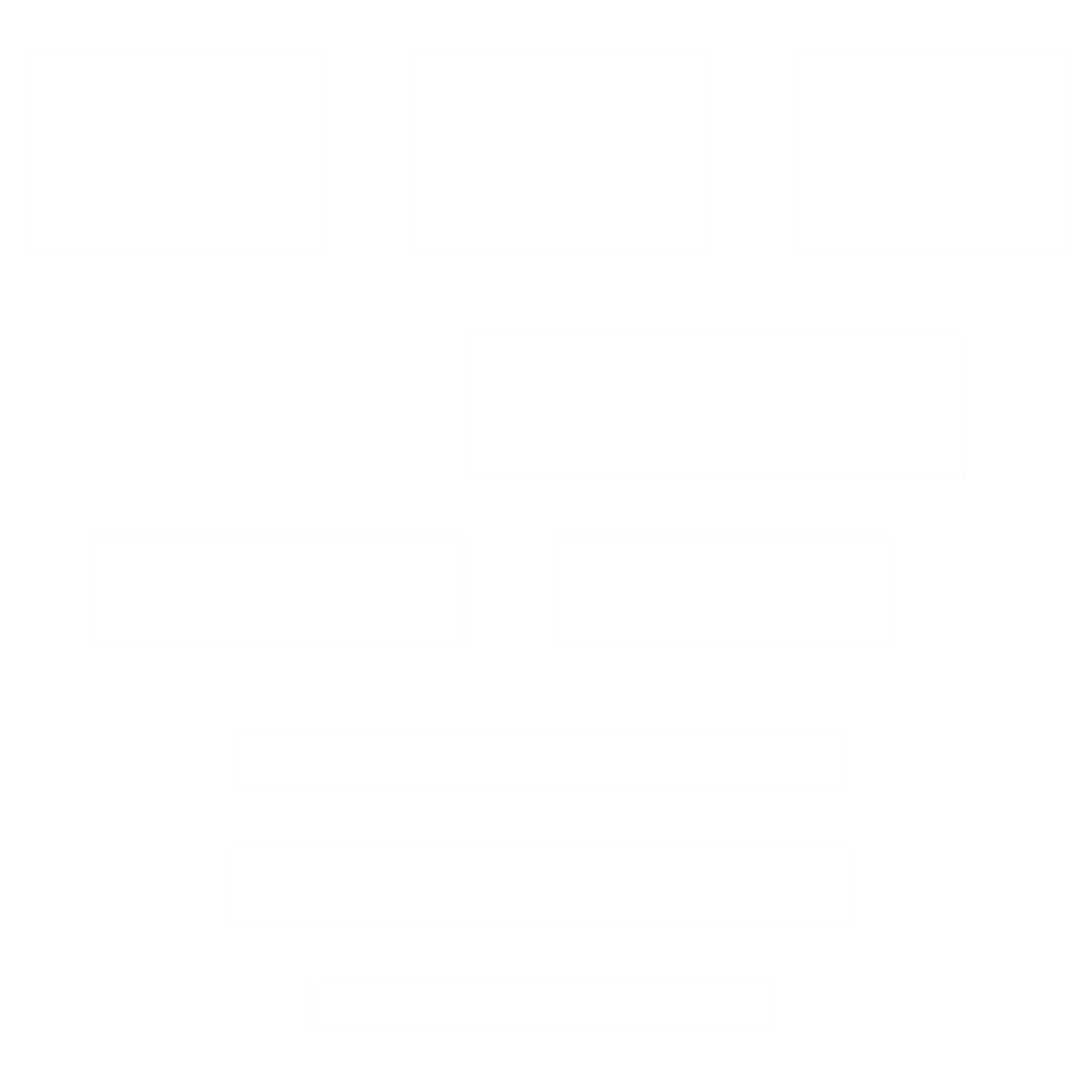
Figure 2: Processus de développement des médicaments biosimilaires. PK = pharmacocinétique ; PD = pharmacodynamique.
Le développement pré-clinique avec l’analyse structurale et fonctionnelle, et clinique des médicaments biosimilaires s’appuie sur la démonstration d’une efficacité et d’une tolérance comparable entre le médicament biosimilaire et le médicament biologique de référence1.
À travers le développement pré-clinique, il s’agit de démontrer une faible variabilité entre la molécule originale et la molécule biosimilaire, sur le plan structural et fonctionnel. Le développement clinique reste spécifique et consiste à s’assurer, une fois la similarité établie, de l’équivalence du rapport efficacité/tolérance en comparaison au médicament de référence. Les effets thérapeutiques à long terme sont identifiés à partir de la pharmacovigilance. Les agences sanitaires considèrent que les effets thérapeutiques du médicament biosimilaire sont similaires à ceux du médicament de référence qui est utilisé depuis de nombreuses années et pour lequel les effets thérapeutiques sont connus.
La rigueur de l’agence européenne qui impose une évaluation de la tolérance et de l’efficacité des médicaments biosimilaires suivant le processus de développement recommandé va donc dans le sens d’une efficacité similaire avec le médicament biologique de référence. Par ailleurs, les dispositifs de surveillance imposés et mis en place par les industriels après la mise sur le marché permettent d’assurer le suivi de l’efficacité et de la sécurité des médicaments biosimilaires.
Il existe actuellement plusieurs catégories de médicaments biosimilaires1 :
Les principaux médicaments développés à ce jour sont ceux dont le brevet est tombé (ou le seront bientôt) dans le domaine public2,3.
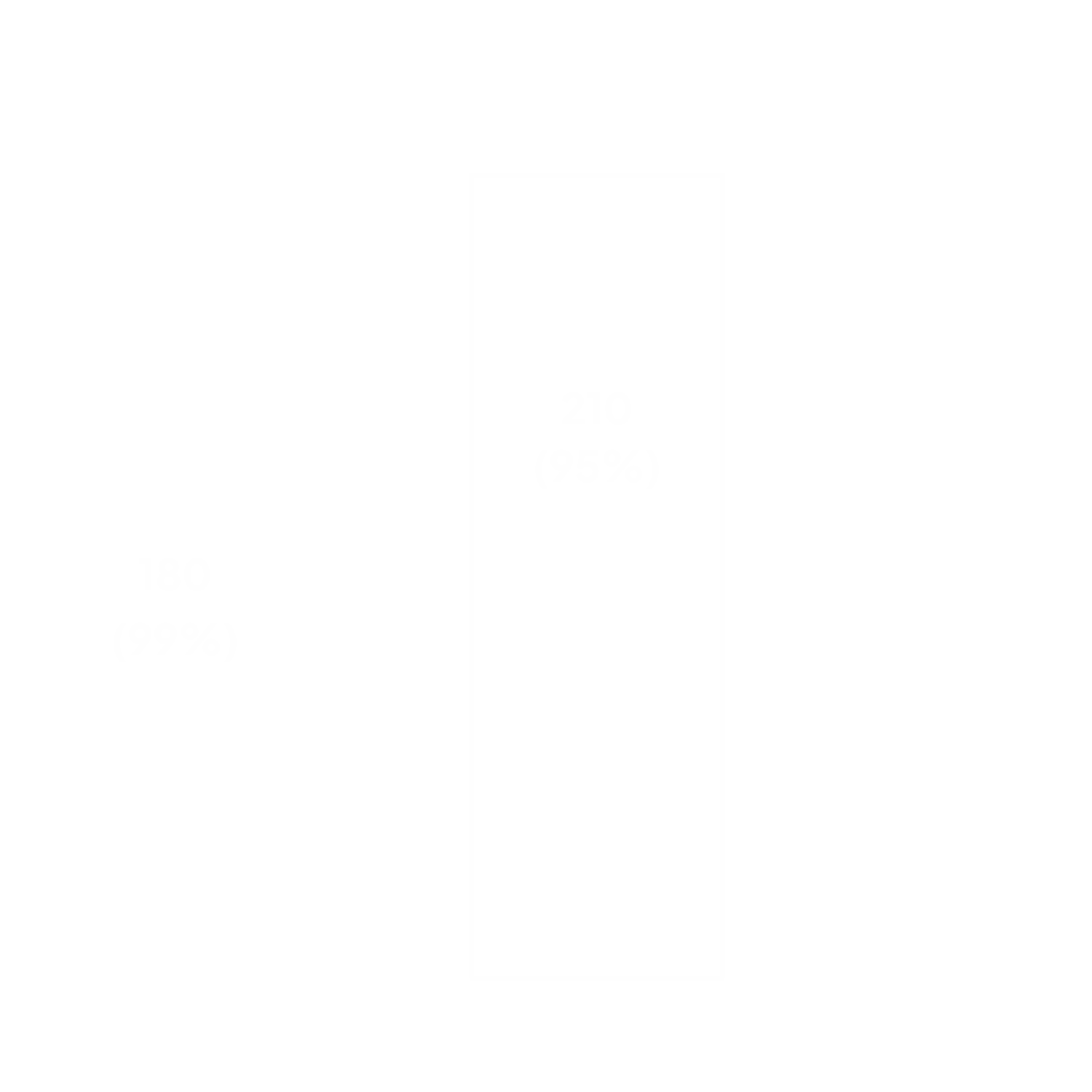
Le marché est donc encore limité, il représentait en 2010 15 % du marché pharmaceutique mondial et devait dépasser 20 % en 2015 avec 200 milliards de dollars 2,3. Comme le montre la figure 4, les prévisions des ventes des produits issus de la biotechnologie augmentent, que ce soient les médicaments biologiques de référence ou les médicaments biosimilaires. La part de ces derniers devrait prendre de l’importance dans les années à venir avec la chute des brevets de plusieurs molécules biologiques très largement prescrites2.
En France, le marché est beaucoup plus ouvert et représente 40 % du marché hospitalier et 25 % du marché pharmaceutique total avec une croissance de 3 à 4 % annuels. Les ventes ont atteint 80 millions d’euros en 2014³.